Synthetic compounds designed to potently and selectively interact with specific biomolecules have served as powerful tools for establishing biological function, and for many biomolecular targets, have resulted in important, life-saving therapeutic agents. The Ellman lab has continually sought to leverage synthetic chemistry to address important biomedical problems. In fact, the design, high throughput/combinatorial synthesis, and testing of drug-like small molecules was the first set of projects to be carried out by the Ellman group, including demonstrating this approach for the first time for nonpolymeric molecules as implemented for the privileged benzodiazepine framework as well as early examples of library design using structure-based docking of virtual libraries with Tack Kuntz (UCSF).
Enzymes are proteins that catalyze a majority of the chemical transformations required by living organisms and as such they play central roles in virtually all biological processes. Enzyme inhibition also provides a powerful approach for the treatment of disease. In collaboration with Charly Craik (UCSF), the Ellman lab developed positional scanning protease substrate profiling methods to rapidly determine preferred cleavage sequences for proteases, enzymes that hydrolyze amide bonds in peptides and proteins. This information can facilitate the identification of natural protease substrates to establish their biological roles and can aid in the design of potent and selective inhibitors. The methods developed with Craik have now been applied to hundreds of proteases by numerous academic and industrial labs.
Due to the rapidity and reliability by which substrate cleavage efficiency can be obtained, the Ellman lab developed a substrate-based fragment identification and optimization approach for small molecule enzyme inhibitor discovery. We first developed the approach as a platform for protease inhibitor discovery and applied it to the identification of potent and selective inhibitors to a number of therapeutically relevant proteases. For example, the Ellman group used the approach to develop highly potent and selective inhibitors of cathepsin S such as 1 (Figure 1A), which is implicated in autoimmune disorders and cancer. Indeed, due to the potency and selectivity of inhibitor 1, it served as the starting point for further drug development by Boehringer Ingelheim, and in collaboration with Matt Bogyo at Stanford, provided the key binding motif for near-infrared activity-based-probes for imaging cathepsin S activity in vivo. Potent, selective and orally available inhibitors such as 2 have also been developed against cruzain (Figure 1B), which is an essential protease of Trypanosoma cruzi and is the causative agent of Chagas’ disease. We have also developed the substrate-based fragment approach for the discovery of small molecule inhibitors to other enzyme classes, including protein tyrosine phosphatases and protein arginine deiminases.
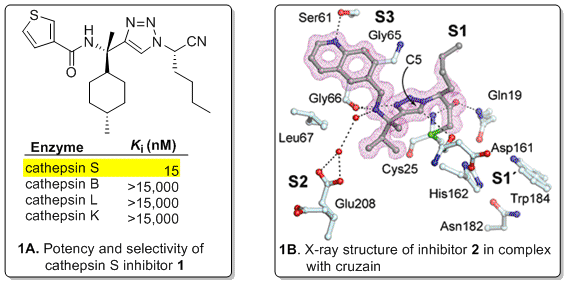
Figure 1. Potent and selective protease inhibitors identified by substrate-based fragment identification and optimization
The depth and breadth of cutting edge research at Yale School of Medicine provides many exciting opportunities for collaboration. As one example, we collaborated with Angus Nairn and Paul Lombroso along with researchers at AZN to identify nanomolar, selective inhibitors of striatal enriched protein tyrosine phosphatase, implicated in neurological disorders, including Alzheimer’s disease (Figure 2), with Ya Ha (Yale Pharmacology) on the structure-based design of potent and selective lipid kinase inhibitors implicated in cancer, and with Anton Bennett, Elias Lolis and Karen Anderson (Yale Pharmacology) on the structure-based design of selective allosteric inhibitors of MKP5, a protein tyrosine phosphatase implicated in diseases such as muscular dystrophy.
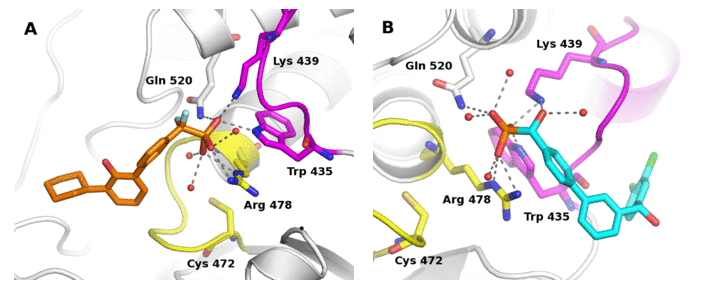
Figure 2. X-ray crystal structures of striatal enriched protein tyrosine phosphatase inhibitors
In collaboration with Brian Shoichet and John Irwin (UCSF) we have generated a virtual library of billions of diverse, druglike non-aromatic nitrogen heterocycles from commercially available inputs based upon C-H functionalization reaction cascades developed by the lab (see C-H Functionalization research section). Through an iterative process of docking, structure-based design, and medicinal chemistry, we have identified potent, selective and pharmacologically active molecules to important therapeutic targets. In one example published in Nature in 2022, we identified potent and selective 5HT2AR agonists that are differentiated from all previously reported 5HT2AR agonists due to their long-acting anti-depressive effects in mouse models, importantly, without eliciting undesired psychedelic effects.