C-H bond functionalization has become one of the most important new methods in synthesis (Figure 1). With an appropriate catalyst, C-H bond functionalization can be performed without a strong acid or base and is highly functional group tolerant thus enabling the efficient synthesis and/or elaboration of drugs and natural products, which typically are rich in functionality. In addition, because virtually every organic compound has C-H bonds, in terms of starting material availability, C-H functionalization approaches theoretically cannot be surpassed.
![]()
Figure 1. Elemental reaction between a C-H bond substrate and a coupling partner
The Bergman and Ellman labs collaborated on the discovery and investigation of new methods centered on catalytic C-H functionalization early in the field’s development, with their first series of publications appearing in 2001. This highly enjoyable collaboration, which continued for more than a decade, also included detailed mechanistic studies of C-H functionalization methods as well as their application to the synthesis of drugs and natural products.
Over the past few years the Ellman lab has explored a new approach for C-H functionalization based upon the three-component sequential coupling of a C-H bond substrate and two different types of coupling partners (Figure 2). This approach provides access to complex structures from simple precursors with the formation of two new bonds. Vast structural diversity is theoretically possible given the enormous number of potential combinations of coupling partners that might be considered
![]()
Figure 2. Basic connection for three-component sequential addition of a C-H bond substrate to two different types of coupling partners
As one example of this type of sequential three-component C-H functionalization reaction, the lab has recently reported on the synthesis of α-branched amines from readily available C-H bond substrates, alkenes, and electrophilic aminating agents (Figure 3). This transformation is highly functional group compatible, can be performed at low catalyst loading, and using chiral catalysts, can be performed in asymmetric fashion.
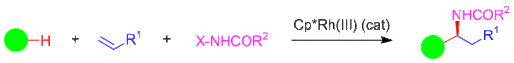
Figure 3. Synthesis of pharmaceutically relevant α-branched amines by Rh(III)-catalyzed three-component coupling of C-H bond substrates, terminal alkenes, and electrophilic aminating reagents
The Ellman lab continues to be highly motivated to develop efficient methods to prepare nitrogen heterocycles, which are present in ~60% of drugs. Annulations proceeding through imidoyl C-H activation is an active area of investigation (Figure 4). Imines, which are easily prepared by condensing large numbers of readily available aldehydes and primary amines, have served as versatile and extremely useful intermediates, though predominantly for nucleophilic additions as exemplified by the well-known Strecker, Mannich, Ugi and Petasis reactions. We believe that imidoyl C-H functionalization for nitrogen heterocycle synthesis opens up a new reaction manifold with the potential to be similarly impactful. Figure 4 depicts select examples of drug relevant heterocycles that we have prepared by catalytic imidoyl C-H activation and annulation using different coupling partners. For some of these transformations, the imines can be prepared in situ enabling straightforward heterocycle synthesis in a single step.
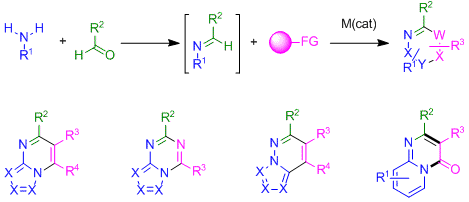
Figure 4. Heterocycle synthesis from readily available precursors by catalytic C-H imidoyl activation and annulation
We have also focused on the use of simple and readily available inputs for the convergent assembly of the piperidine framework, the most prevalent nitrogen heterocycle in drugs. Rh(I)-catalyzed C-H alkenylation followed by electrocyclization provides 1,2-dihydropyridines, which in the same pot or after simple filtration, can then be transformed into highly functionalized piperidine derivatives (Figure 5). Heterocycles from low to high levels of substitution as well as polycyclic derivatives can readily be accessed using this approach.
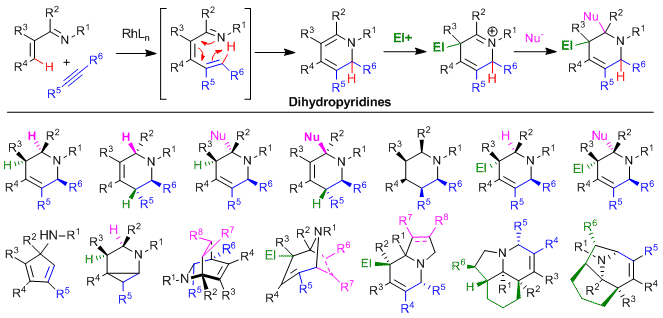
Figure 5. Convergent synthesis of piperidines by C-H bond functionalization cascade processes
When new reactions are developed, we investigate their mechanisms through the use of isotope labeling, kinetic analysis, and x-ray structural characterization of catalyst resting states and intermediates. Figure 6 depicts representative structures of metal complexes that have been characterized in the context of mechanistic inquiry.
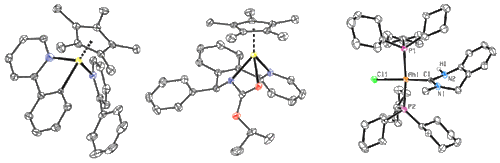
Figure 6. Examples of x-ray structures of transition metal complexes from mechanistic studies
As we develop new C-H bond functionalization methods, we also apply them to the synthesis of relevant natural products and drugs (Figure 7). This includes the first total synthesis of (+)-lithospermic acid and the first total synthesis of the semi-synthetic opioid antidote (-)-naltrexone.
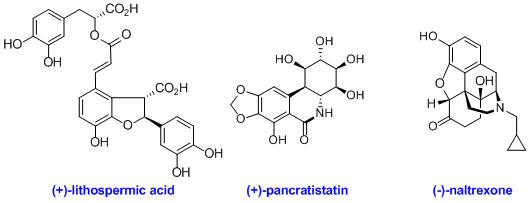
Figure 7. Representative natural products and drugs synthesized using C-H bond functionalization